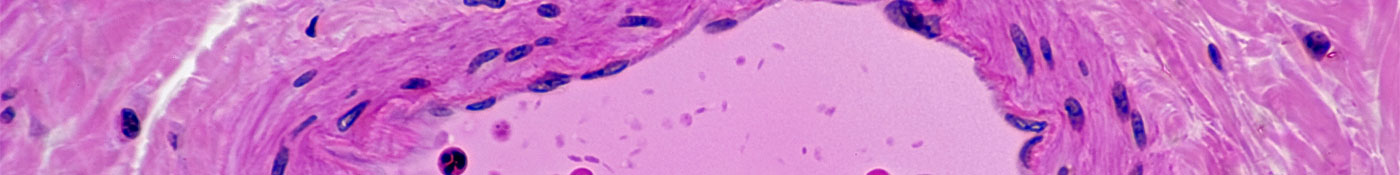
Construimos nuestro futuro legado y extendemos nuestra experiencia para transformar la PAH en una
afección crónica controlable.

La PAH afecta a las personas de todas las edades, altera la vida cotidiana y limita la supervivencia. Nuestro objetivo inmediato es prolongar la supervivencia y también queremos mejorar la calidad de vida de aquellos que tienen esta enfermedad.
La PAH se presenta en personas de todas las edades, incluso niños; sin embargo, los pacientes son diagnosticados de PAH cuando tienen entre 50 y 65 años. 1,2 Los síntomas de PAH, como falta de aire, fatiga, dolor en el pecho y sensación de mareo, tienen un gran impacto en el funcionamiento y el bienestar físico, psicológico y social de los pacientes.3
En promedio demora dos años desde la aparición de los síntomas para que se diagnostique la PAH. 4 y muchos pacientes pueden no ser identificados.5 A pesar de los avances recientes, la PH es una enfermedad grave y progresiva que no tiene cura.6,7
Mediante programas de investigación y descubrimiento compartidos en todas las áreas de tratamiento, estamos destinando importantes recursos de descubrimiento para revelar los mecanismos más prometedores que pueden modificar el curso de la PAH. Nuestro objetivo es revertir las patologías preexistentes en la PAH y resolver la enfermedad.
Para lograrlo, estamos accediendo a nuestra profunda experiencia in vitro e in vivo y conectándonos con las comunidades de pacientes e de investigadores para comprender completamente los desafíos y los riesgos asociados a la PAH.
Otros tipos de hipertensión pulmonar
Las investigaciones hasta el momento se han enfocado principalmente en la PAH, pero hemos ampliado nuestra investigación para incluir la hipertensión pulmonar tromboembólica crónica (CTEPH, por sus siglas en inglés) y otros tipos de PH.
Referencias
1. Galiè N, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2016; 37:67-119.
2. Hoeper MG and Gibbs SR. The changing landscape of pulmonary arterial hypertension and implications for patient care. Eur Respir Rev 2014; 23:450-7.
3. Chin KM, et al. Psychometric Validation of the Pulmonary Arterial Hypertension-Symptoms and Impact (PAH-SYMPACT) Questionnaire. Results of the SYMPHONY Trial. Chest 2018: 154(4):848-61.
4. Prins KW, et al. WHO Group I Pulmonary Hypertension: Epidemiology and Pathophysiology. Cardiol Clin 2016; 34(3):363-74.
5. Humbert M, et al. Pulmonary Arterial Hypertension in France. Results from a National Registry. Am J Respir Crit Care Med 2006; 173:1023-30.
6. Vachiéry JL and Gaine S. Challenges in the diagnosis and treatment of pulmonary arterial hypertension. Eur Respir Rev 2012; 21:313-20.
7. Hoeper MM, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J 2017; 50:1700740.
Pulmonary Hypertension
Pulmonary Hypertension