

Pulmonary Hypertension
Pulmonary Hypertension
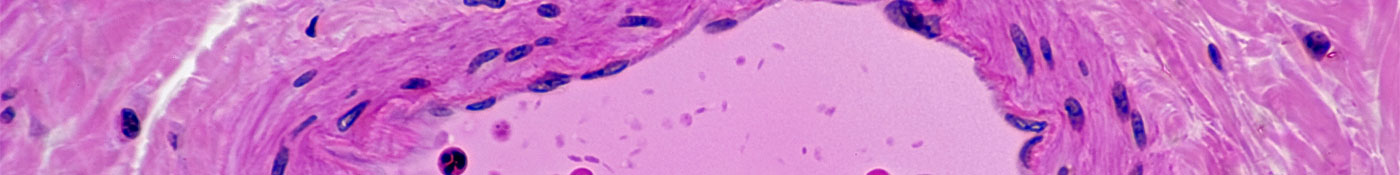
Hipertensão arterial pulmonar (HAP) é uma doença fatal, mas pode ser controlada—se você receber um diagnóstico precoce. A seguir, estão as informações que as pesquisas científicas mais recentes revelaram sobre a HAP, incluindo o principal grupo de risco e os sinais de alerta a que de se deve atentar.
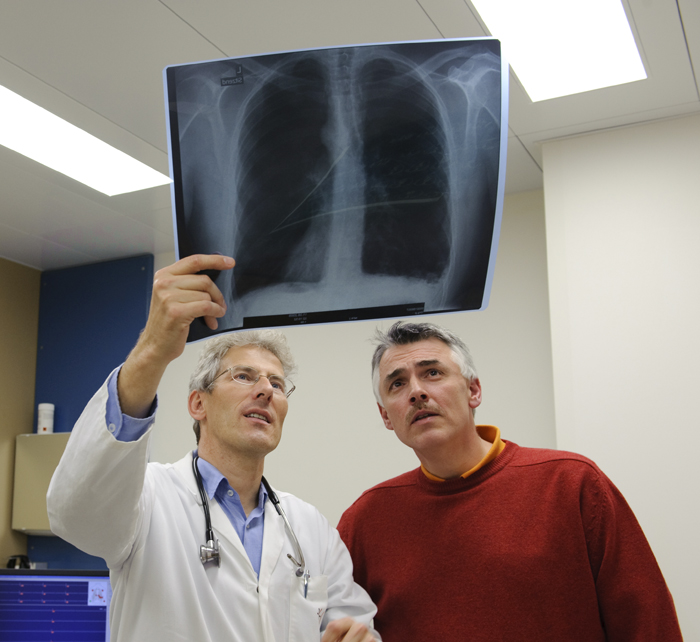
A hipertensão é tão comum—cerca de um em três adultos americanos a possui—que é provável que você conheça alguém com esta doença. Pode ser que você mesmo a tenha.
Por outro lado, a hipertensão arterial pulmonar é muito mais rara—e é potencialmente muito mais perigosa.
Quando alguém é diagnosticado com a hipertensão normal (sistêmica), isso significa que o sangue realiza uma pressão muito forte contra as paredes das artérias, aumentando os riscos de ataque cardíaco e infarto.
Hipertensão pulmonar é um tipo de alta pressão sanguínea que ocorre nos pulmões. Ela pode ser ocasionado por uma variedade de fatores, como apneia do sono e doença pulmonar obstrutiva crônica, conhecida como DPOC. A hipertensão arterial pulmonar (HAP) é um dos tipos de hipertensão pulmonar, ocorrendo quando as paredes das artérias que saem do lado direito do coração para os pulmões se estreitam e ficam apertadas. Como resultado, a pressão nos pulmões aumenta, gerando sintomas como fatiga e falta de ar.
Embora a HAP seja uma doença rara—sua real prevalência ainda é desconhecida, mas estima-se que entre 50 a 100 pessoas por milhão possuam a doença—receber o diagnóstico e o tratamento corretos é de extrema importância para quem tem a doença, pois a HAP piora com o tempo, e atualmente é incurável.
Se você está dentro deste grupo, ou apenas está curioso(a) para compreender mais sobre esta condição frequentemente fatal, aqui está uma cartilha sobre a HAP.
1. A causa da HAP é desconhecida, mas mudanças fisiológicas são fundamentais para seu desenvolvimento e progressão.
A HAP é diagnosticada quando a pressão sanguínea média das artérias pulmonares for medida em 25 mmHg ou mais. Ninguém sabe o motivo exato por trás da corrente de eventos que ocasionam a HAP, mas especialistas sabem muito sobre seu progresso.
“Ela se inicia com a constrição de uma dessas artérias, levando a um remodelamento de suas paredes”, explica Alessandro Maresta, M.D., Vice President and Head of Global Medical Affairs da Actelion Pharmaceuticals, uma companhia de biotecnologia suíça que a Johnson & Johnson adquiriu em junho de 2017. “Conforme as paredes das artérias engrossam, o lúmen—o espaço por onde o sangue flui—diminui e a pressão aumenta. Com a progressão da doença, a pressão continua a subir, mas o ventrículo direito cardíaco não é projetado para bombear sangue contra uma pressão tão alta.”
À medida que a doença se agrava, “os pacientes costumam ficar muito cansados e sentir falta de ar”, afirma Nazzareno Galié, M.D., professor de cardiologia e Director of the Specialization School of Cardiology, Universidade de Bologna, Itália. “Algumas pessoas podem notar palpitações cardíacas ou sentir pressão no peito, especialmente ao se exercitar”.
2. Há quatro tipos de HAP.
De acordo com Galié, “a maioria dos casos são considerados idiopáticos”, ou seja, não há uma causa conhecida ou fator precipitante de maior importância.
Cerca de seis por cento dos casos são hereditários. Eles são herdados de um dos pais, provavelmente causados por uma mutação em um receptor proteico que afeta o crescimento tecidual. “Caso esteja carregando este gene, não significa que você desenvolverá HAP, mas que está correndo um maior risco,” adverte Maresta. “Acreditamos que é necessário mais de um fator desencadeador,” como uma condição pré-existente.
As formas idiopáticas e hereditárias da HAP são, no mínimo, duas vezes mais comum em mulheres do que em homens. De fato, HAP é mais frequentemente diagnosticada em mulheres com idade entre
30 e 60 anos.
Em alguns casos, a HAP pode ser causada por fármacos. O medicamento mais famoso por causar HAP é a combinação de fenfluramina e fentermina, um remédio para perda de peso popular nos anos 1960 e 1970, mas que não está mais a venda.
Por fim, casos associativos ocorrem quando a HAP é concomitante com outra condição pré-existente, como HIV, doença congênita cardíaca ou escleroderma.
Todos os quatro tipos—idiopático, hereditário, causado por fármacos e associativo—são tratados de forma semelhante,mas o prognóstico pode variar de acordo com o tipo.
Por exemplo, observa Maresta, pacientes que possuem HAP associativa porque também sofrem de escleroderma são mais propensos a desenvolverem doenças renais, complicações gastrointestinais e cardíacas. “Isso pode impactar sua expectativa geral de vida”, conclui Maresta.
3. A HAP afeta mais as mulheres do que os homens.
As formas idiopáticas e hereditárias da HAP são, no mínimo, duas vezes mais comum em mulheres do que em homens, de acordo com a Pulmonary Hypertension Association. De fato, HAP é mais frequentemente diagnosticada em mulheres com idade entre 30 e 60 anos.

Atualmente, desconhece-se o motivo pelo qual mulheres são mais suscetíveis, mas algumas teorias sob investigação apontam para o papel do estrogênio, mudanças (incluindo as hormonais) que ocorrem durante a gravidez e conexões com doenças autoimunes, que também são mais prevalentes em mulheres.
4. A HAP é de difícil diagnóstico.
Dada a sua raridade, quando um paciente queixa-se de sintomas como exaustão e respiração forçada, o mais usual é que o médico primeiramente realize testes para detectar doenças como asma e insuficiência cardíaca congestiva.
Embora faça mais sentido de uma certa maneira—essas doenças são muito mais comuns do que a HAP, explica Gailé,—também significa que ser diagnosticado com HAP pode, às vezes, demorar um certo tempo.
“Você pode se consultar um clínico geral, então um especialista para asma, então outro especialista e assim em diante, até finalmente ver um pneumologista ou cardiologista que então diagnosticam a HAP”, elucida Maresta. “Geralmente demora um ano entre o início dos sintomas e o diagnóstico correto e final de HAP”.
Além disso, observa Gailé, “seu médico também precisará descartar todas as outras formas de hipertensão pulmonar antes de finalizar o diagnóstico em HAP”.
Mas a boa notícia é que o tempo até o diagnóstico de HAP está começando a acelerar um pouco. “As coisas estão mudando aos poucos, assim que mais médicos sabem sobre HAP e como identificá-la”, explica Maresta.
Uma vez que seu médico suspeita de HAP, ele (provavelmente um cardiologista ou pneumologista) pedirá uma série de testes, que podem incluir um raio-X do peito, teste de função pulmonar e de tolerância ao exercício. Se todos os sinais apontarem para a HAP após os resultados dos testes, você provavelmente precisará de um cateterismo do lado direito do coração, que mede a pressão dentro das artérias pulmonares.
5. Apesar de não haver cura para a HAP, há maneiras efetivas de controlar a doença.
Em um nível psicológico, há diferentes mecanismos biológicos—conhecidas como vias—relacionadas a como vasos sanguíneos funcionam, que acredita-se desempenharem um papel na HAP.
“Segundo as diretrizes, é altamente recomendável que, quando um paciente for diagnosticado com HAP, ele comecem a tomar uma combinação de remédios que têm como alvo duas vias distintas,” afirma Maresta. “Você pode receber um remédio primeiro e, em seguida, começar o segundo dentro de seis meses”. Maresta acrescenta que estudos atuais estão sugerindo que atingir três vias ao mesmo tempo, possui o potencial de gerar melhores resultados.
“A média de sobrevida (desde o diagnóstico) costumava ser de dois anos e meio. Agora, eu diria que a maioria dos pacientes está vivendo de sete a dez anos, e alguns chegam a sobreviver até vinte anos.”
Alessandro Maresta. M.D.
Vice President and Head of Global Medical Affairs, Actelion Pharmaceuticals
Embora não haja uma cura atualmente para a HAP, o prognóstico típico é muito melhor hoje do que há 25 anos. “A média de sobrevida (desde o diagnóstico) costumava ser de dois anos e meio”, diz Maresta. “Agora, eu diria que a maioria dos pacientes está vivendo de sete a dez anos, e alguns chegam a sobreviver até vinte anos.”
O foco principal atual, acrescenta Maresta, é encontrar uma nova via e um fármaco efetivo para atingi-la—para tanto há pesquisas atualmente em curso.
“Estamos nos dedicando para tornar isso realidade”, afirma Maresta.
Este artigo, escrito por Barbara Brody, foi publicado pela primeira vez no www.jnj.com.