Construindo nosso futuro legado e expandindo nossa experiência para transformar a HAP em uma
condição crônica e tratável.

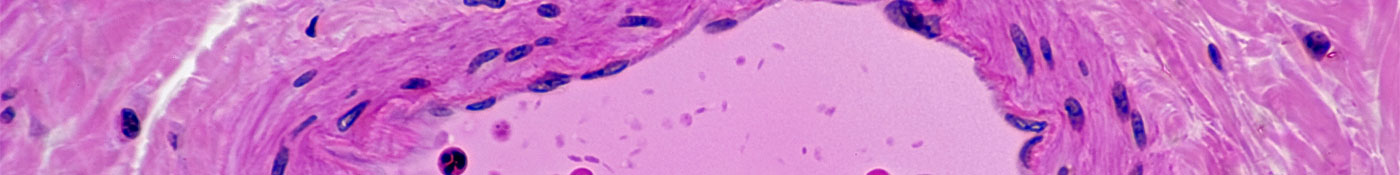
A HAP afeta pessoas de todas as idades, prejudicando o dia a dia e limitando a sobrevivência. Nosso objetivo imediato é prolongar a sobrevida e também melhorar a qualidade de vida das pessoas que têm essa doença.
A HAP pode ocorrer em todas as idades, incluindo em crianças, no entanto, a maioria dos pacientes com HAP são diagnosticados entre 50 e 65 anos de idade.1,2 Os sintomas de HAP, como falta de ar, fadiga, dor no peito e tontura, têm um importante impacto no bem-estar funcional, físico, psicológico e social do paciente.3
Em média, são necessários dois anos desde o início dos sintomas até que a HAP seja diagnosticada,4 e muitos pacientes podem nem receber o diagnóstico.5 Apesar dos avanços recentes, a HAP é uma doença grave e progressiva, sem cura.6,7
Com programas de pesquisa e descoberta compartilhados em nossas áreas terapêuticas, estamos investindo recursos significativos para descobrir os mecanismos mais promissores que podem modificar o curso da HAP. O nosso objetivo é reverter as patologias subjacentes da HAP e resolver a doença em si.
Para isso, estamos aproveitando nossa ampla experiência in vitro e in vivo, além de nos conectar a comunidades de pacientes e pesquisadores para compreender totalmente os desafios e riscos associados à HAP.
Outras formas da hipertensão pulmonar
Até o momento, as pesquisas têm se concentrado principalmente na HAP, mas estendemos nossa pesquisa para incluir a hipertensão pulmonar tromboembólica crônica (CTEPH) e outros tipos de HP.
Referências
1. Galiè N, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2016; 37:67-119
2. Hoeper MG and Gibbs SR. The changing landscape of pulmonary arterial hypertension and implications for patient care. Eur Respir Rev 2014; 23:450-7.
3. Chin KM, et al. Psychometric Validation of the Pulmonary Arterial Hypertension-Symptoms and Impact (PAH-SYMPACT) Questionnaire. Results of the SYMPHONY Trial. Chest 2018: 154(4):848-61.
4. Prins KW, et al. WHO Group I Pulmonary Hypertension: Epidemiology and Pathophysiology. Cardiol Clin 2016; 34(3):363-74.
5. Humbert M, et al. Pulmonary Arterial Hypertension in France. Results from a National Registry. Am J Respir Crit Care Med 2006; 173:1023-30.
6. Vachiéry JL and Gaine S. Challenges in the diagnosis and treatment of pulmonary arterial hypertension. Eur Respir Rev 2012; 21:313-20.
7. Hoeper MM, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J 2017; 50:1700740.
Pulmonary Hypertension
Pulmonary Hypertension