

Lungenhochdruck
Lungenhochdruck (Pulmonal arterielle Hypertonie/PAH) – Krankheit, Symptome, Diagnose und Behandlung
 ein weitgehend normales Leben führen können.")
Ziel ist es, dass Menschen mit PAH (Lungenhochdruck) ein weitgehend normales Leben führen können.
Was ist Lungenhochdruck (pulmonale Hypertonie)?
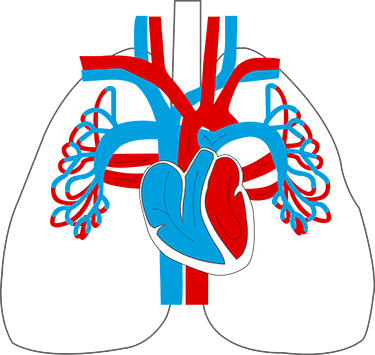
Bei Lungenhochdruck (pulmonale Hypertonie; kurz PH) sind die Blutgefäße der Lunge verengt. Sind speziell die Arterien betroffen, also die vom Herzen wegführenden Gefäße, spricht man von einer pulmonal arteriellen Hypertonie (PAH).
In Folge der Verengung steht der kleine Kreislauf zwischen Lunge und Herz (Lungenkreislauf) unter permanent erhöhtem Druck.
In diesem Kreislauf wird das sauerstoffarme Blut von der rechten Herzkammer in die Lunge gepumpt, wo es wieder mit Sauerstoff angereichert wird und dann von der linken Herzkammer erneut in den großen Kreislauf fließt. Sind nun die Lungengefäße verengt, muss die rechte Herzkammer verstärkt arbeiten, um genügend Blut durch die Gefäße zu pumpen.
Die Verengung der Gefäße wird durch ein Ungleichgewicht der körpereigenen Botenstoffe ausgelöst. Dabei wird der Botenstoff Endothelin, die stärkste bekannte gefäßverengende Substanz im Körper, vermehrt produziert. Gleichzeitig kommen die gefäßerweiternden Gegenspieler seltener vor.
Anzeichen und Symptome der PAH
Erste Symptome von Lungenhochdruck sind Beeinträchtigungen bei körperlichen Tätigkeiten wie Treppen steigen oder Gepäcktragen.
Dazu gehören:
- Luftnot
- schnelle Erschöpfung
- Müdigkeit
- Schwindel
- Herzrasen
Auch ein Gefühl der Brustenge kann auftreten. Da diese Symptome auch auf andere Ursachen zurückzuführen sind, ist eine Diagnose im frühen Stadium schwer. Erst mit dem Fortschreiten der Krankheit werden die Symptome deutlicher:
- weiterhin abnehmende Belastbarkeit und zunehmende Luftnot
- Ohnmachtsanfälle während oder nach körperlicher Belastung
- Herzklopfen, Herzrasen
- starke Brustschmerzen, ähnlich wie bei einem Herzinfarkt
Ist die rechte Herzkammer betroffen, treten zudem folgende Symptome auf:
- Wasseransammlungen in den Beinen, gestaute Halsvenen – Blaufärbung der Lippen
- Reizhusten, evtl. mit Blutbeimengung
- niedriger Blutdruck im Körperkreislauf
Diagnose der PAH
Eine frühe Diagnose von PAH ist schwer. Besteht ein Verdacht, ist es wichtig, sich an eine:n Lungenspezialist:in oder Kardiolog:in zu wenden. Die ersten Symptome lassen bestenfalls einen Verdacht zu und sind oftmals auch auf andere Ursachen zurückzuführen. Untersuchungen wie EKG oder ein Röntgen von Herz und Lunge zeigen zu Anfang ebenso oftmals keinen Befund.
Um eine Diagnose stellen zu können, wird neben einem Bluttest und weiteren spezifischen Untersuchungen von Herz und Lunge (Goldstandard ist der Rechtsherzkatheter) auch die körperliche Belastbarkeit getestet. Liegt eine Diagnose vor, sind weitere Untersuchungen notwendig, um die Ursache und den Stand der Erkrankung gezielt festzustellen und die richtige Therapie zu wählen.
Leben mit PAH: Behandlung und Verlauf
Bei PAH handelt es sich um eine chronische Krankheit, die das Leben der Betroffenen nachhaltig beeinflusst. PAH ist zwar nicht heilbar, allerdings gibt es inzwischen, anders als vor 25 Jahren, verschiedene Behandlungsmöglichkeiten. Dank großer Fortschritte in der Forschung stehen Betroffenen heute wirksame Therapien zur Verbesserung der Sauerstoffversorgung durch Erweiterung der Blutgefäße und der körperlichen Leistungsfähigkeit zur Verfügung.
Ärzt:innen entscheiden über die Therapie individuell. In der Regel werden in der Behandlung der PAH eingesetzt:
- allgemeine Maßnahmen wie kontrollierte körperliche Aktivität
- Basistherapeutika
- spezifisch wirkende Medikamente
Da die Ursachen des Lungenhochdrucks verschieden sein können, ist auch die Therapie der einzelnen Patient:innen sehr individuell.
Zu den Basistherapien können die Sauerstoffgabe, die Behandlung mit Gerinnungshemmern und entwässernde Medikamente gehören. Diese sollen vor allem die Symptome lindern und so zu einer Verbesserung des Allgemeinbefindens beitragen. Die spezifisch wirkenden Medikamente können drei unterschiedliche Signalwege im Körper adressieren. Allen ist gemein, dass sie durch eine Erweiterung der Gefäße zu einer Entlastung des Herzens beitragen. Verschiedene Präparate können mitunter auch kombiniert werden, mit dem Ziel, die Wirkung zu verbessern und genau den Bedürfnissen der einzelnen Patient:innen zu entsprechen. Je nach Stadium der Erkrankung können Betroffene mit der passenden Therapie ein weitgehend normales Leben führen.
Häufige Fragen zur PAH
Was sind Risikofaktoren für eine PAH?
Grundsätzlich kann jede:r an einer PAH erkranken. Es gibt allerdings einige Risikogruppen: Dazu zählen insbesondere Patient:innen mit Sklerodermie (systemischer Sklerose), Menschen mit einem angeborenen Herzfehler (auch wenn dieser korrigiert wurde) sowie HIV-Patient:innen.
Ist PAH vererbbar?
Nur eine Form ist vererbbar und diese kommt recht selten vor: die hereditäre pulmonal-arterielle Hypertonie (hPAH).1 Die hPAH beruht am häufigsten auf der Mutation eines bestimmten Gens, das BMPR2 genannt wird. BMPR2 steht für „Bone-morphogenetic-protein-Rezeptor 2“ und gehört zur Familie der transformierenden Wachstumsfaktoren TGF-Beta. Bei anderen Patient:innen können seltenere Mutationen vorliegen, bei der ebenfalls bestimmte Wachstumsfaktoren der TGF-Beta-Familie betroffen sind.2 Wachstumsfaktoren spielen eine wichtige Rolle bei der Entwicklung von Zellen und Geweben. Für eine genetische Beratung sollte man sich am besten an eine:n Lungenhochdruckexpert:in werden.
Wie häufig ist PAH?
Registerdaten aus wirtschaftlich entwickelten Ländern gehen von 48 bis 55 Fällen pro einer Million Erwachsener aus.3 Für Deutschland würde dies bedeuten, dass rund 4.000 bis 4.700 Erwachsene an PAH erkrankt sind. Da die Diagnose der seltenen Krankheit oft schwierig ist und häufig erst nach Jahren gestellt wird, kann die Dunkelziffer auch deutlich höher sein.
Ist eine Schwangerschaft mit PAH möglich?
Eine Schwangerschaft mit PAH ist sowohl für Mutter als auch Kind lebensgefährlich. Bestimmte Medikamente gegen die Krankheit können zudem Missbildungen beim Fötus verursachen. Daher sollten Patientinnen mit einem Kinderwunsch unbedingt im Voraus mit ihren Ärzt:innen sprechen.
Welche körperliche Aktivität ist bei PAH möglich?
Das ist von Person zu Person verschieden. Keine Bewegung ist allerdings eine schlechte Option. Zu empfehlen ist ein moderates Training unter Anleitung und Aufsicht von Spezialist:innen. Spezielle Dehn- und Kräftigungsübungen lassen sich zum Beispiel im Rahmen der Krankengymnastik erlernen, um das Wohlbefinden ganzheitlich zu verbessern.
Weitere Angebote für Patient:innen und Interessierte
Janssen With Me
Weiterführende Informationen zu Symptomen, Diagnose und Therapieoptionen bei Lungenhochdruck.
mein heute mein morgen
Hinter dem Service für Patient:innen steht ein fachkompetentes Team, das bei sämtlichen erkrankungsbezogenen Themen helfen kann.
Lungeninformationsdienst
Verständlich aufbereitete, aktuelle und wissenschaftlich geprüfte Information aus allen Bereichen der Lungenforschung und Medizin.
Informationen für medizinische Fachkreise
Janssen Medical Cloud
Weiterführende Informationen zu Ursachen, Symptomen und Diagnose von pulmonaler Hypertonie.
Die Informationen, die wir Patient:innen zur Verfügung stellen, können einen Besuch bei Ärzt:innen nicht ersetzen. Zudem können wir aus gesetzlichen Gründen Patient:innen und sonstigen Laien im Sinne des Heilmittelwerberechts (§ 10 HWG) keine werblichen Informationen über verschreibungspflichtige Medikamente bereitstellen.
- Lungeninformationsdienst: Lungenhochdruck: Was ist das? https://www.lungeninformationsdienst.de/krankheiten/lungenhochdruck/grun.... Letzter Zugriff am 08.02.2024
- Montani D et al. Pulmonary arterial hypertension. Orphanet J Rare Dis. 2013; 8:97. https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-8-97. Letzter Zugriff am 08.02.2024
- Humbert M et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2022; 43(38): 3618–3731. https://pubmed.ncbi.nlm.nih.gov/36017548/. Letzter Zugriff am 08.02.2024
EM-150586