Pulmonary arterial hypertension (PAH) is a deadly disease, but it can be managed—if you get a timely diagnosis. Here is what the latest science and research has revealed about PAH, including who is most at risk and the warning signs to look out for.

High blood pressure, otherwise known as hypertension, is so common—about one in three American adults has it—that you likely know someone with the condition. You might even have it yourself.
Pulmonary arterial hypertension, on the other hand, is much more rare—and can be potentially much more deadly.
When someone is diagnosed with regular (systemic) hypertension, it means that the force of blood pushing against the sides of the arteries is too strong, which raises the risk of heart attack and stroke.
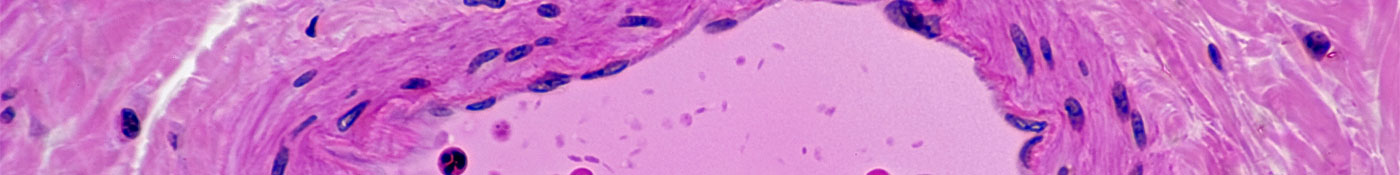
Pulmonary hypertension is a form of high blood pressure that occurs in the lungs. It can be triggered by a variety of factors, including sleep apnea and chronic obstructive pulmonary disease or COPD, as it's commonly called. PAH is one type of pulmonary hypertension, which occurs when the walls of the arteries leading from the right side of the heart to the lungs narrow and tighten. As a result, pressure in the lungs rises, leading to symptoms like fatigue and shortness of breath.
Although PAH is a rare disease—true prevalence is still unknown, but it's estimated that between 50-100 people per million have the condition—getting the right diagnosis and treatment is crucial for those who have it because PAH worsens over time, and there's currently no cure.
Whether you're among that group or you're just curious to understand more about the often deadly condition, here's a primer on the facts about PAH.
1. The exact cause of PAH is unknown, but physiological changes are key to its development and progression.
PAH is diagnosed when mean, or average, blood pressure leaving the arteries on the right side of the heart is measured at 25 mmHg or higher. No one knows exactly what kicks off the chain of events that sets PAH in motion, but experts do know a lot about how it progresses.
"It starts with constriction of these vessels and leads to remodeling of the vessel wall," says Alessandro Maresta, M.D., Vice President and Head of Global Medical Affairs for Actelion Pharmaceuticals, a Swiss biotech company that Johnson & Johnson acquired in June 2017. "As the walls of the blood vessels thicken, the lumen—the space where blood flows through—gets smaller, and the pressure goes up. As the disease progresses, pressure continues to rise, but the right ventricle of the heart is not designed to pump blood against such high pressure."
As the condition worsens, "patients tend to become very tired and short of breath," says Nazzareno Galié, M.D., Professor of Cardiology and Director of the Specialization School of Cardiology, University of Bologna, Italy. "Some people might notice heart palpitations or feel chest pressure, especially when exercising."
2. There are four types of PAH.
According to Galié, "most types are considered idiopathic," meaning there's no known cause or major precipitating factor.
About six percent of cases are heritable. They're passed down from a parent, most likely due to a mutation in a protein receptor that affects tissue growth. "If you're carrying this gene it does not mean you're definitely going to develop PAH, but you are at a higher risk," Maresta says. "We think you need more than one trigger," such as a pre-existing condition.
The idiopathic and heritable forms of PAH are at least twice as common in women compared to men.
In fact, PAH is most often diagnosed in women between the ages of 30 and 60.
In some cases, PAH can be drug-induced. The medication most famous for causing PAH is fen-phen (fenfluramine and phentermine), a weight loss drug that was popular in the 1960s and 1970s but is no longer on the market.
Lastly, associative cases occur when PAH goes hand in hand with another pre-existing condition, such as HIV, congenital heart disease or scleroderma.
All four types—idiopathic, heritable, drug-induced and associative—are treated similarly, but the prognosis might vary as a result of which one you have.
For example, notes Maresta, patients who have associative PAH because they also have scleroderma are already more prone to renal disease, gastrointestinal complications and heart complications. "That can impact your overall life expectancy," Maresta adds.
3. PAH affects women more than men.
The idiopathic and heritable forms of PAH are at least twice as common in women compared to men, according to the Pulmonary Hypertension Association. In fact, PAH is most often diagnosed in women between the ages of 30 and 60.

It's not currently known why females are more susceptible, but some theories under investigation have to do with the role of estrogen, changes (including hormonal ones) that occur during pregnancy and links to autoimmune issues, which also tend to be more prevalent in women.
4. PAH is difficult to diagnose.
Given how rare PAH is, when a patient complains about such symptoms as exhaustion and labored breathing, it's not unusual for a doctor to first order testing for conditions like asthma and congestive heart failure.
While that makes sense on some level—those issues are much more common than PAH, explains Galié—it also means that getting diagnosed with PAH may sometimes take a while.
"You might see a general practitioner, then a specialist for asthma, then another specialist and so on before finally seeing a pulmonologist or cardiologist who diagnoses the PAH," Maresta explains. "It usually takes one year from the presentation of symptoms to get a correct and final diagnosis of PAH."
Plus, notes Galié, "your doctor will also need to rule out all other forms of pulmonary hypertension before giving a diagnosis of PAH."
But the good news is that the timeline to PAH diagnosis is starting to speed up a little. "Things are slowly changing, as more doctors now know what PAH is and how to identify it," Maresta explains.
Once your physician suspects PAH, your doctor (likely a cardiologist or pulmonologist) will order a series of tests that may include a chest X-ray, pulmonary function test and exercise tolerance test. If all signs still point to PAH following the results of these tests, you'll probably require a right-heart catheterization, which measures the pressure inside the pulmonary arteries.
5. While there’s no cure for PAH, there are effective ways to manage the disease.
On a physiological level, there are several different biological mechanisms—known as pathways—related to how blood vessels work that are believed to play a role in PAH.
"The guidelines strongly recommend that when a patient is diagnosed with PAH, they start a combination of drugs to target two different pathways," Maresta says. "You might get one drug first, and then start on the second one within six months." Maresta adds that current studies are now suggesting that targeting three pathways at once has the potential to lead to better outcomes.
"The median survival [from time of diagnosis] used to be two and a half years. Now I'd say most patients are living seven to 10 years, and some are living as long as 20 years."
Alessandro Maresta. M.D.
Vice President and Head of Global Medical Affairs, Actelion Pharmaceuticals
While there's currently no cure for PAH, the typical prognosis is much better today than it was 25 years ago. "The median survival [from time of diagnosis] used to be 2.5 years," Maresta says. "Now I'd say most patients are living seven to 10 years, and some are living as long as 20 years."
The main focus today, adds Maresta, is to find a new pathway and an effective drug to target it—for which research is currently underway.
"We're dedicated to making it happen," Maresta says.
This article, written by Barbara Brody, first appeared on www.jnj.com.
Pulmonary Hypertension
Pulmonary Hypertension